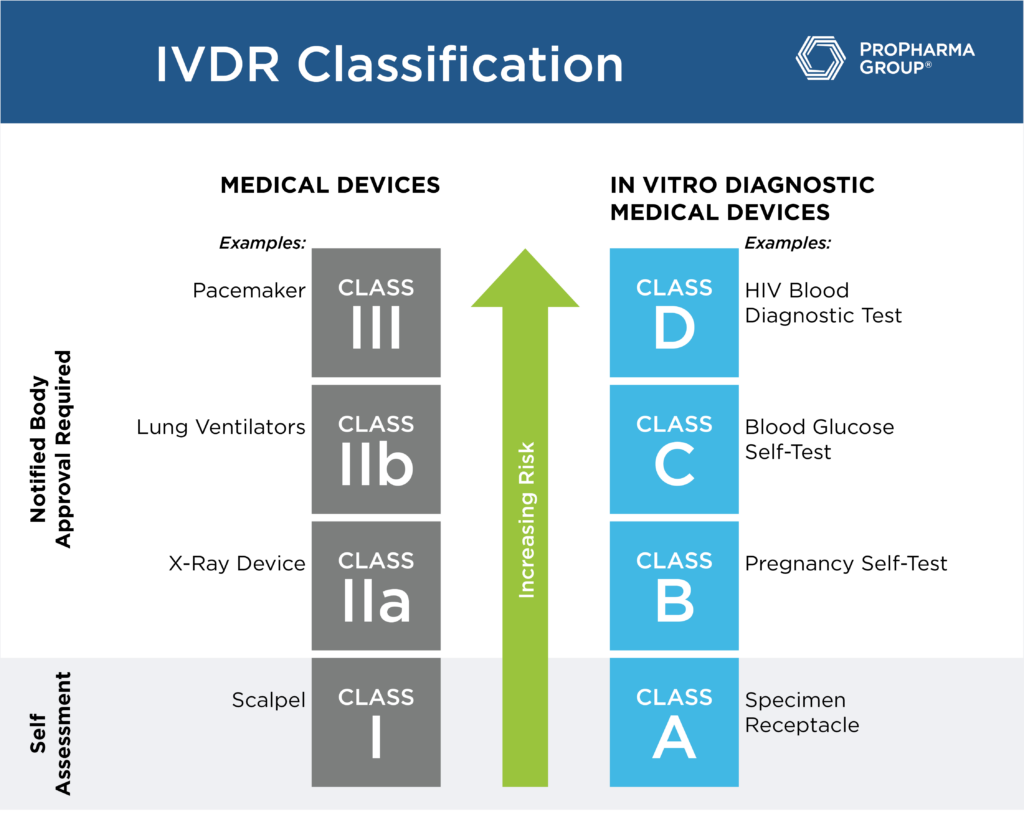
With an increased need for high quality in vitro diagnostic medical devices (IVDs), the In Vitro Diagnostic Regulation (IVDR, 2017/746) was entered into force for all IVDs in 2017 with a five-year transition period. The end of this transition period is May 2022, leaving manufacturers with very little time before their technical documentation needs to be updated to comply with the regulations and meet the new, more stringent requirements. This blog will answer the top critical questions, and more information about Medical Devices & Diagnostics can be found here.
What will be the impact of this change for manufacturers with products in the EU and what do we expect for US markets? Having a comprehensive and efficient strategy will help you ensure a rapid and successful pathway to market access. Here are answers to the top critical questions to prepare for the new regulation.
Will the deadline of May 2022 be extended?
No
Even with the delay in some products and having them fully certified, what needs to be done by May 2022, regardless of products getting their CE mark.
The quality management adjustments should be made as soon as possible, per article 10, paragraph 8. Performance evaluation should be addressed to the IVDR level ASAP as well.
What do the new Eudamed modules mean for me? When do you use it, how do you transition to it without work arounds?
Still some time to go before going live for other parties than the NB and CAs. No workaround necessary at this time.
How can my department prepare for this transaction?
As referenced in article 10 and paragraph 8, start updating the technical documentation to the IVDR level sooner rather than later.
Which digital trends will IVDR changes fuel the most?
Don’t forget thorough software validation according to the standards, and don’t forget about GDPR.
Is there a need to prove safety and performance as per MEDDEV 2.7.4.? What other general testing is required to demonstrate performance?
The MEDDEV 2.7. series is applicable to medical devices, not IVDs. But yes, some GSPR must be addressed by the performance evaluation process.
How have you helped other organizations who have struggled with EU IVDR or MDR projects? How do you translate your expertise to being able to tangibly support or address our needs?
We have conducted gap assessment projects to understand and prioritize what needs to be done to address IVDR/MDR compliance. We have also provided subject matter experts to run mock audits or address specific needs regarding IVDR/MDR compliance (e.g., PMSS, Clinical or Performance Compliance). Finally, we have provided overarching project management support to successfully oversee and drive these programs to completion.
Is ISO 20916 applicable for clinical performance studies, instead of ISO 14155?
Yes, that is correct!
If you have a question we didn’t answer here, connect with our team and learn more about our solutions to successfully prepare for the new regulations.