July 2, 2024

July 2, 2024
As of January 31, 2023, Sponsors are required to submit a Clinical Trial Application (CTA) in the EU through the Clinical Trial Information System (CTIS). This EU portal is established as part of the new Clinical Trial Regulation No. 536/2014 (CTR) which harmonizes and centralizes the submission and assessment of Clinical Trial Applications in the EU. The CTR is applicable to clinical trials with Gene Therapy Medicinal Products (GTMPs) as well, but unfortunately it does not cover the environmental requirements that are applicable to GTMPs if they contain or consist of genetically modified organisms (GMOs). In this respect, a clinical trial with a GMO must also comply with either the contained use Directive (2009/41/EC) or the deliberate release Directive (2001/18/EC). The Directive that actually applies to a clinical trial with GMOs is dependent upon the individual Member States (MS) (see Figure 1).
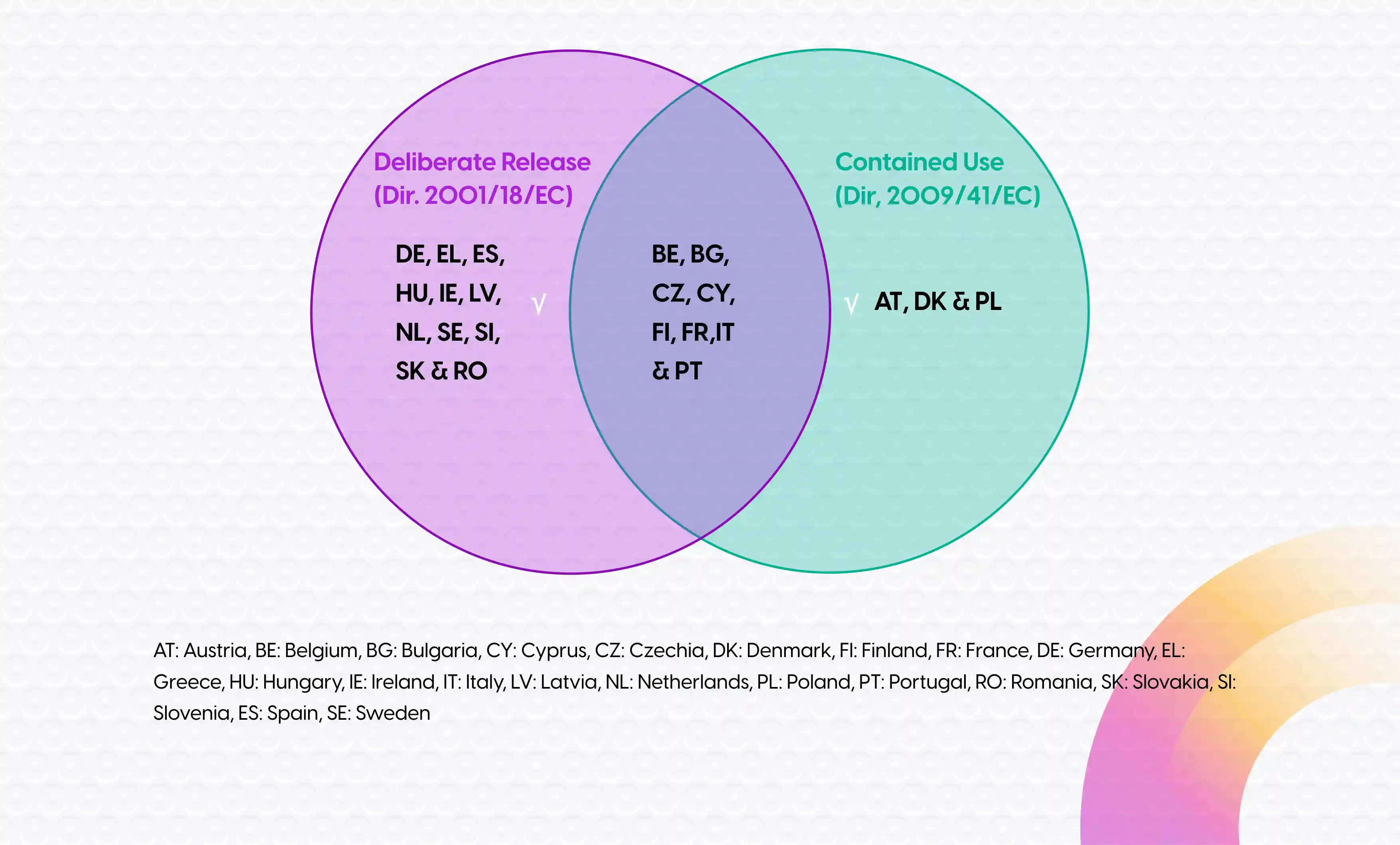
Regardless of the applicable Directive, a GMO application must be submitted to each MS of choice in addition to the CTA. As discussed in our previous blog , for most clinical trials with GTMPs a Common Application Form (CAF) is the central document of the GMO application. However, if the clinical trial is considered to be deliberate release by the MS, the GMO application also needs to include a Summary Notification Information Format (SNIF) form1.
Until recently, this SNIF form had to be submitted together with the other GMO documents to the Competent Authority (CA) of each Member State involved in the clinical trial. However, as of October 2022, the submission process has changed and the SNIF form is now required to be submitted separately from the GMO application through the E-Submission Food Chain (ESFC) platform.
As stipulated in Article 11 of Directive (2001/18/EC), information about the deliberate release in the environment of GMOs, like clinical trials with GTMPs, need to be shared with all Member States in the EU. In line with this European notification procedure, a public summary should accompany the GMO application of a clinical trial with GTMPs. The required information for this public summary is defined by a so-called Summary Notification Information Format. This SNIF form needs to be completed and, until recently, sent to the Competent Authority (CA) of the MS(s) involved in the clinical trial together with the other GMO documents, e.g. common application form, environmental risk assessment, etc.
The CA receiving the GMO application has always been responsible for the exchange of the SNIF information with the European Commission (EC). With the enforcement of the Transparency Regulation (EU/2019/1381), this process has been adjusted. While the GMO application still needs to be sent to the Competent Authority of the particular MS, the SNIF form should be sent to the EC directly via the ESFC platform. Subsequently, the involved CA needs to assess the SNIF within the ESFC platform. Although this change is seemingly minor and the platform is readily accessible after creating a login account, it doesn’t ease the burden that is often felt around the GMO application either. In fact, it adds a new layer to the process and this process is not the same in all MS.
In principle, using the ESFC platform to submit the SNIF, while submitting the remainder of the GMO application directly to the CA of the involved MS, doesn’t seem too complicated. In this respect, it should be noted that the SNIF form is populated within the platform and cannot be prepared offline and uploaded as a document. Furthermore, although it is suggested that both the SNIF and the GMO application can be submitted in parallel, this is not necessarily the case and the order in which these can be submitted, does vary between MS. For example, in Germany, the SNIF information has to be submitted to the ESFC platform first and proof of this submission has to be added to the GMO application. On the other hand, in Spain, the entire GMO application, including the SNIF (in Spanish), needs to be submitted to the Ministry of Agriculture, Food and Fisheries first. Only after their approval, the SNIF (in English) is to be submitted to the ESFC platform.
Moreover, it is unclear how to deal with amendments to the CTA which affect the SNIF as well. This was previously managed by the CA but is now the responsibility of the Sponsor. The current service desk of the ESFC platform does not seem to be sufficiently equipped to clarify any content-related concerns. As a result, a simple addition of clinical sites in a Member State can become quite cumbersome.
Sponsors need to understand the ESFC platform and need to acquire information regarding the differences between Member States in the submission process of the SNIF form. However, submission of the SNIF form through the ESFC platform should not be problematic for Sponsors that are active in the field of gene therapy trials and accustomed to the European environmental legislation that applies to the use of GTMPs. Nevertheless, it does not ease the process around the GMO application either and can be considered an additional layer of complexity.
So, the question remains: why has it been decided to introduce a different platform for the submission of the SNIF form in times where the CTIS was introduced as central location for EU wide CTA submissions? Wasn’t it an ideal opportunity to include the submission of the SNIF through CTIS? Obviously, this would be superfluous in case a MS is involved, which considers a clinical trial with a GTMP as contained use instead of deliberate release. However, in this respect, Part II of the CTA dossier for CTIS, which is focused on MS specific information seems an ideal location for the SNIF form.
Apparently, it was not feasible yet to take this small step towards harmonization and centralization of the GMO and CTA applications, which is definitively needed to make the EU more attractive for clinical trials with GTMPs. In this respect, the proposal for new pharmaceutical legislation2 that was adopted on the 26th of April 2023 by the EC, might relieve some of the burden by proposing the possibility to submit the environmental risk assessment or CAF through CTIS. Hopefully, this includes the possibility to submit the SNIF form through CTIS also.
ProPharma has dominant knowledge and skills within the regulatory world for cell and gene therapies and is uniquely positioned to help you navigate these requirements. We have significant expertise with regards to GMO requirements and submissions and are continually reviewing the regulatory landscape to maintain current knowledge of the latest legislative developments within the cell and gene therapy realm to best support our Clients. We regularly work with clients preparing applications and can provide any support that you might need, from advice regarding the contained use or deliberate release procedure and the required documents for each of these procedures, to the preparation, submission and management of required GMO applications across all territories required
TAGS: EU Europe General Regulatory Regulatory Sciences Cell and Gene Therapy

July 1, 2024
The Benefit of the Common Application Form for GMO Applications GMO Application Not Included in Clinical Trial Regulation With the transition period for the Clinical Trial Regulation No 536/2014...

March 26, 2024
On April 26, 2023, the European Commission proposed a new package of pharmaceutical legislation1 to revise many of the currently applicable Regulations. Background This revision is considered by many...

October 31, 2024
On January 31, 2022, the EU Clinical Trials Regulation 536/2014 (CTR) came into force with a transition period of three years. Now, as that transition period comes to a close, the process and need...