
The Benefit of the Common Application Form for GMO Applications
GMO Application Not Included in Clinical Trial Regulation
With the transition period for the Clinical Trial Regulation No 536/2014 (CTR) complete, as of January 31, 2023, it is now mandatory to use the centralized Clinical Trial Information System (CTIS) for submission of all clinical trial applications (CTAs) within the European Union (EU). Although this regulation applies to CTAs for gene therapy studies, it does not cover the additional requirement for a Genetically Modified Organism (GMO) application for such studies. This lack of covering the GMO applications is considered a missed opportunity since the requirements, procedures, and timelines for a GMO application vary greatly between Member States. Therefore, despite the promised harmonization CTR brings to authorization of clinical trials in Europe, for gene therapy trials the application remains cumbersome and time-consuming, especially for multicenter trials in different Member States.
Fortunately, specific common application forms (CAF) for Gene Therapy Medicinal Products (GTMPs) have been launched, which ease the GMO application preparation for gene therapy trials in Europe to a certain extent.
The Complex Regulatory Requirements for a GMO Application
In contrast to other investigational medical products, most gene therapy medicinal products contain or consist of genetically modified organisms. Consequently, a GTMP clinical trial is subjected to specific environmental legislation and requires a GMO application in addition to the commonly known CTA. In this respect, two different Directives, in addition to the CTR, can apply to a GTMP clinical trial in the EU:
- Directive 2001/18/EC on deliberate release
- Directive 2009/41/EC on contained use
Whether a clinical trial with GTMPs is seen as deliberate release or contained use differs between EU Member States. Moreover, each Member State has implemented both Directives into national legislation leading to diverging requirements and imposition of different measures for protecting contact with the trial subject and the environment. Additionally, each Member State has their own procedures and timelines for the assessment of the GMO application. Therefore, significant knowledge, time, and resources are required to apply for a multicenter GTMP clinical trial in several Member States. Especially, considering the comparatively straightforward procedure in the United States (US), in which a categorial exclusion will suffice in most cases.
Three Different Common Application Forms for Clinical Trials for GTMPs
To ease the burden of the discordant GMO application process in the EU, a good practice document1 2, was developed covering the assessment of GMO-related aspects in the context of clinical trials with adeno-associated virus (AAV) vectors and with genetically modified human cells. Based on the current experience with clinical trials with either AAV-vectors or genetically modified human cells, the good practice document includes a standardized environmental risk assessment, which is applicable to most trials with these types of GMOs. As a result, separate CAFs for AAV-vectors3 and for genetically modified human cells4 have been generated to be used as central document for the GMO application. In this form only a limited set of information about the GMO and its clinical use is requested and it can be used in every Member State that endorsed the relevant CAF. Unfortunately, these forms haven’t been endorsed by all Member States and the Member States that endorsed the CAF for AAV-based trials didn’t necessarily endorse the CAF for trials with genetically modified human cells and vice versa (see Figure 1, below). Nevertheless, a good number of Member States did accept these forms, including those that have hosted the most GTMP trials in recent years.
Figure 1: Endorsement Common Application Forms by different EU Member States
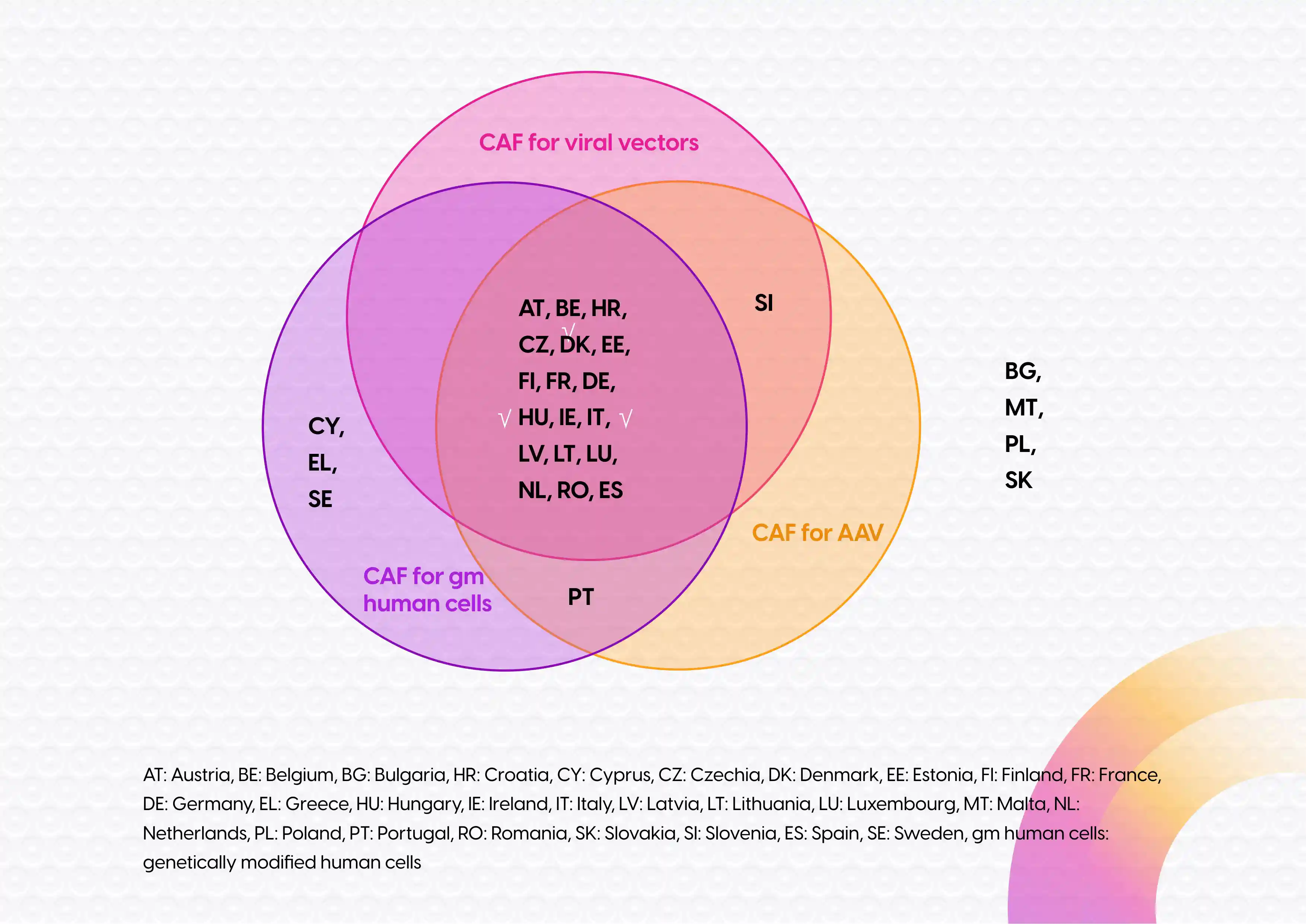
In addition, a third CAF has been generated that is generally applicable for clinical trials with viral vectors (excluding AAV-vectors)5 . In contrast to the abovementioned forms, this CAF is not based upon a good practice document with a standardized environmental risk assessment. Consequently, a GMO application for a clinical trial with a viral vector still requires an environmental risk assessment, which should be provided within the CAF.
Benefits of Common Application Form (CAF) for GTMPs
The benefit of the CAF is that just one central document is to be prepared for a multicenter GTMP trial in different Member States, although some minor differences in the requirements between the Member States remain and require some fine-tuning is needed regarding the information provided.
Next-Level Streamlining Approach in the Netherlands
In the Netherlands, in case of clinical trials with AAV vectors and with human cells genetically modified by a lentiviral or retroviral vector, the approach has been pushed even further. This is because clinical sites are defined as GMO license holders instead of the Sponsor. Ahead of future clinical trials, many of the main Dutch medical centers involved in the conduct of clinical GTMP studies have applied for a general GMO license for either AAV-based trials or trials with genetically modified human cells, or even both. The Dutch Competent Authority has granted these platform GMO licenses for clinical trials with both types of GTMPs. Consequently, a trial-specific GMO application is not required for studies involving these licensed medical centers, leaving 'just’ the submission of a CTA via CTIS. This Dutch approach can be considered a step towards alignment with, or even improvement on the process of categorial exclusion in the US.
CTR Brings Regulatory Harmonization in the EU
The CTR harmonizes the CTA within the EU but does not ease the burden of complex GMO applications required for clinical trials with GTMPs. Fortunately, the development of a CAF for clinical trials with AAV-based vectors and genetically modified human cells is a step towards simplifying the GMO application for GTMP studies. Moreover, in the Netherlands, several medical centers now have a platform GMO license in place for gene therapy studies based on these two types of GTMPs, which can negate the requirement for a separate GMO application completely.
Although, the implementation of the CAF eases the legislative burden for GMO applications to a certain extent, differences in requirements and procedures between the different Member States in the EU still remain. It is hoped that further advances will be implemented, to make the EU more attractive for the conduct of GTMP clinical studies now and in the future. In this respect, the proposal for a new pharmaceutical legislation6 that was adopted on April 26, 2023 by the EC, is of particular interest. In this proposal, amendments of, among others, the Clinical Trial Regulation are suggested and include the possibility to submit the environmental risk assessment or CAF via the EU portal CTIS and the subsequent assessment by the Committee for Medicinal Products for Human Use.
ProPharma: Your Expert Regulatory Affairs Consultant
ProPharma has dominant knowledge and skills within the regulatory world for cell and gene therapies and is uniquely positioned to help you navigate these requirements. We have significant expertise with regards to GMO requirements and submissions and are continually reviewing the regulatory landscape to maintain current knowledge of the latest legislative developments within the cell and gene therapy realm to best support our Clients. We regularly work with clients preparing applications and can provide any support that you might need, from advice regarding the contained use or deliberate release procedure and the required documents for each of these procedures, to the preparation, submission and management of required GMO applications across all territories required.
References
- Good Practice on the assessment of GMO related aspects in the context of clinical trials with AAV clinical vectors; Good Practice document AAV vectors
- Good Practice on the assessment of GMO-related aspects in the context of clinical trials with human cells genetically modified; Good Practice document genetically modified human cells
- Common application form for investigational medicinal products for human use that contain or consists of AAV vectors; CAF AAV vectors.
- Common application form for clinical research with human cells genetically modified; CAF genetically modified human cells.
- Common application form for viral vectors contained in investigational medicinal products for human use; Common Application Form for viral vectors.
- Proposal for a Regulation of the European Parliament and of the Council; Proposal for new pharmaceutical legislation.
TAGS: Europe General Regulatory Regulatory Sciences Cell and Gene Therapy (CAGT/CGT)


