July 16, 2024

July 16, 2024
The key to successful drug development in the US is directional and focused navigation of FDA’s Investigational New Drug (IND) process. The Chemistry, Manufacturing, and Controls (CMC) section is a critical component of an IND submission and is integral to US regulatory applications. The data provided in the CMC sections within Module 3 of an IND application provide FDA with information on the drug’s:
Although there are FDA regulatory guidelines regarding the minimum CMC content requirements, in reality no two Module 3 sections of an IND are alike. The IND Module 3 content is driven by the specific science and technology of the product and process along with the goals of the drug development program.
The quantity and level of detail of information provided in the CMC section varies depending on the product’s stage of development. As the Sponsor’s knowledge and manufacturing process and control experience increases, so do FDA’s expectations of the details and supporting data generated and reported during drug development. If done properly, and with frequent communication with the FDA, this will result in a complete and comprehensive set of CMC information that could eventually be used to support product approval and commercialization.
Figure 1, below, illustrates FDA’s general expectations regarding the level of CMC product and process activities, increasing as product advances through the stages of drug development.
Figure 1: Increasing CMC/Process Knowledge Based on Phase of Development
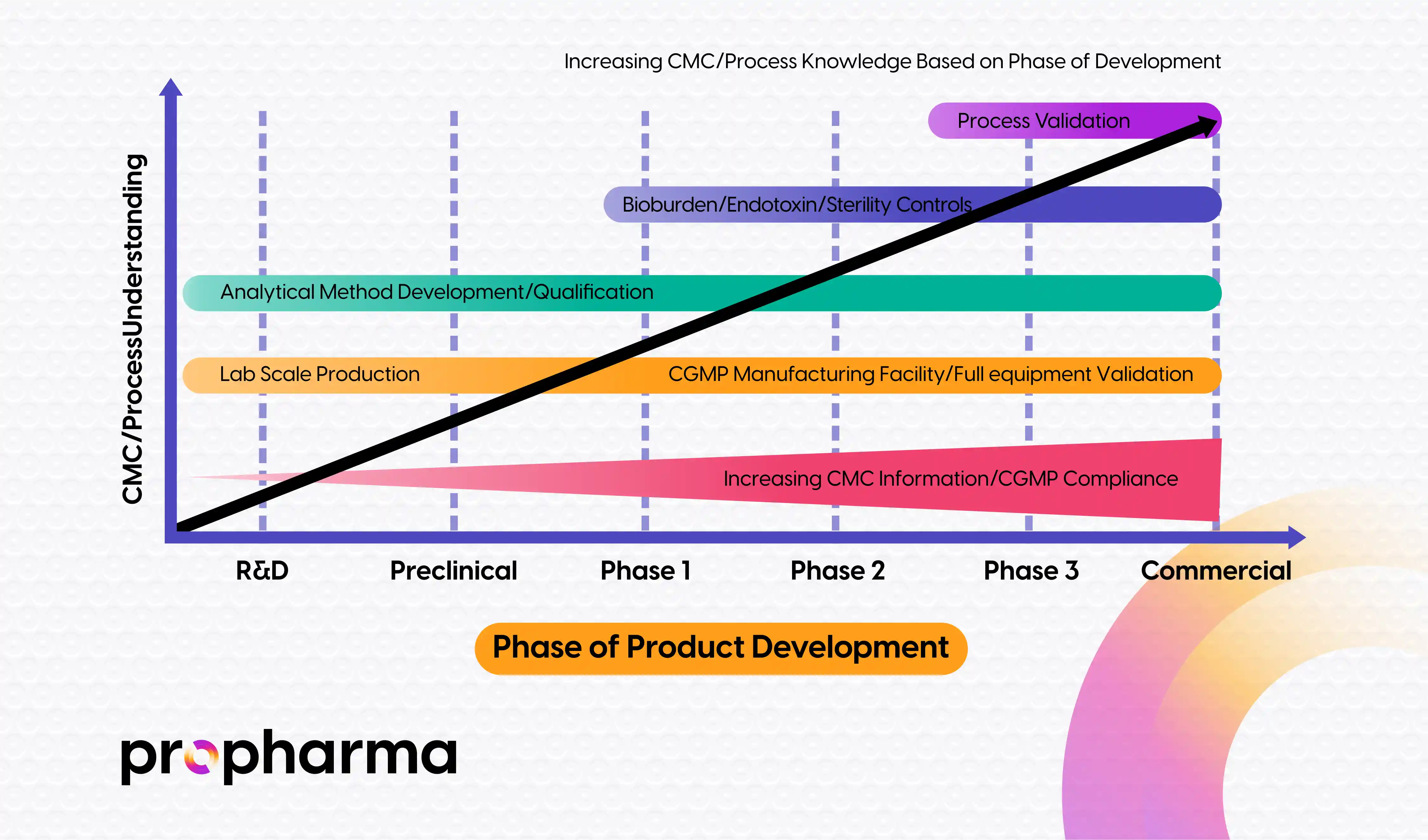
At the Pre-IND stage of development, Sponsors have identified the target drug molecules’ synthetic scheme and have developed preliminary drug substance (DS) and drug product (DP) manufacturing processes. Typically, at this stage, the DS has been manufactured at lab scale, and characterization experiments have been conducted to establish its identity and purity. Preliminary stability under accelerated or stressed conditions would be considered and applied to gain an understanding of the number and approximate levels of impurities that may arise during the manufacturing process and storage. Preliminary analytical methods would be developed and partially qualified to inform the quality of the manufactured product. Impurity levels would be expected to be monitored to comply with the International Council for Harmonisation of Technical Requirements of Pharmaceutical for Human Use (ICH) expectations and toxicological safety studies may be assessed and/or conducted as needed.
Once sufficient information has been obtained from lab scale manufacturing processes, the DS and DP processes are then transitioned and or transferred to GMP-compliant facilities for common Good Manufacturing Practice (CGMP) manufacturing of clinical trial materials. Characterization and CMC information are again collected (as stated above, however the information gathered at this stage is now more detailed and data rich) and compiled into an initial Module 3 of an IND submission to support the five CMC Pillars of an IND, which include:
Additional details on the five CMC pillars of an IND submission are illustrated in Figure 2, below.
Figure 2: The Five CMC Pillars of a Drug IND
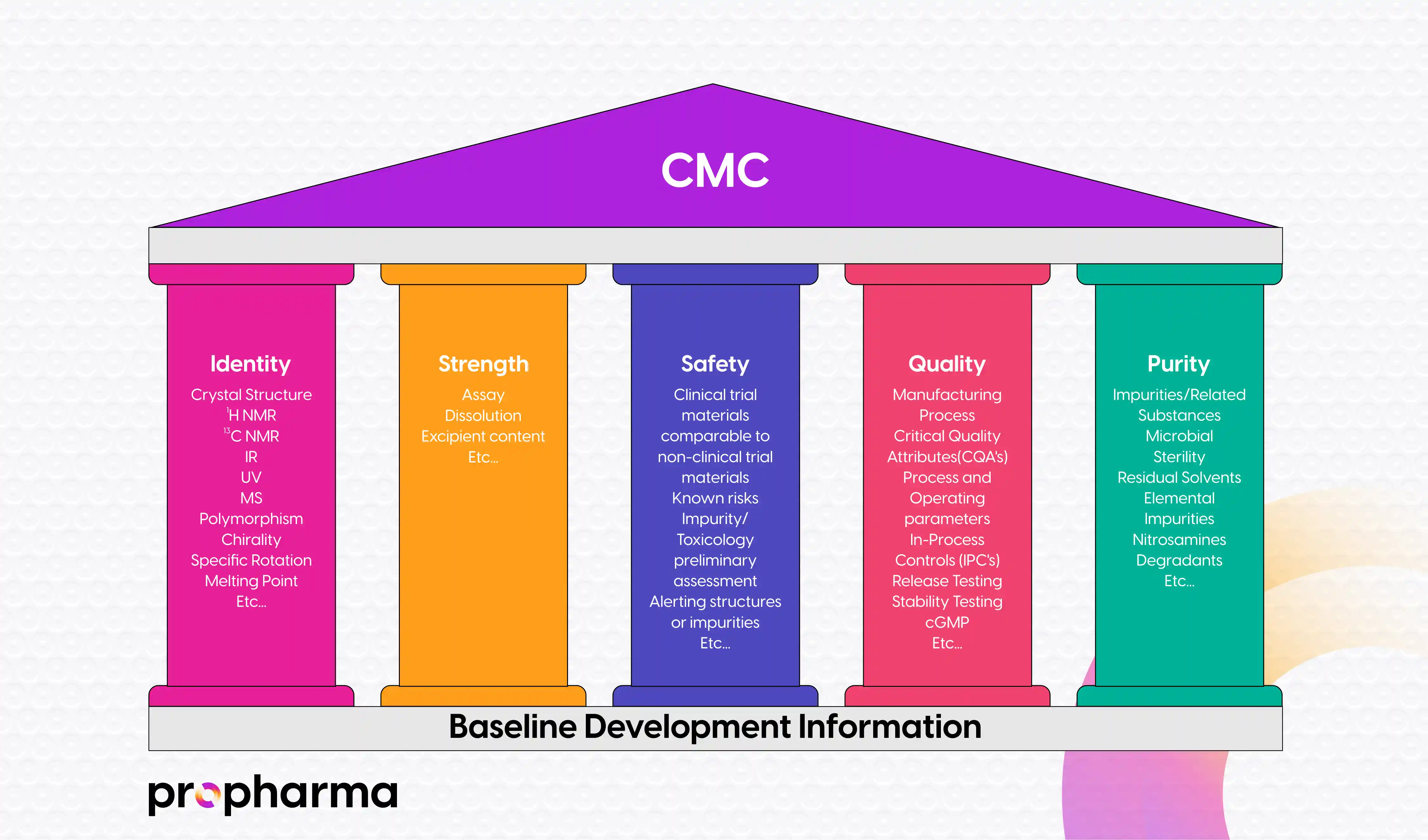
During the process of compiling an IND submission, additional CMC development knowledge and understanding is also gained. Changes at this stage could include:
Finally, comparability and clinical bridging must be contemplated, risk assessed, and executed as applicable. The IND submission should be updated throughout the clinical trial phases, incorporating this new CMC knowledge and understanding. Frequent updates and communications with FDA are highly encouraged to mitigate surprises at the Phase 3 and NDA stages.
The FDA End of Phase 2 milestone becomes critically important for the CMC program. In addition to establishing the Phase 3 clinical requirements, Sponsors can clarify with the FDA specific CMC areas and or CMC filing strategies (e.g., minimum stability data required, adequacy of two registration batches with three primary stability batches at the time of NDA filing, etc.) that will enable a successful FDA NDA submission.
Validation questions, methods and acceptance criteria, extractable/leachable evaluations, process expansion plans, and lockdown of the route of administration and container closure system should all happen at this time. Doing these tasks during the EOP2 will ensure the Phase 3 pivotal clinical materials are representative of the proposed commercial processes and product.
Prior to submitting an FDA NDA submission, Sponsors should be prepared with the following:
When all of the items listed above are complete, all of the CMC information, supporting data, and reports should then be organized and summarized in the applicable respective eCTD Module 3 sections of the NDA filing:
The key to successful drug development and approval in the US is the directional and focused navigation of the FDA IND process. When done correctly and in conjunction with regular advice and guidance from the FDA, Sponsors will have a complete and comprehensive set of CMC information that is supportive of FDA approval and product commercialization in the US.

June 25, 2024
Recently, the FDA updated a long-standing, decades old guidance on analytical test method validation based on revisions of the ICH Q2(R2) guidelines. Traditional test method validation requirements...

June 24, 2025
For drug developers leveraging the 505(b)(2) pathway, the FDA Pre-Investigational New Drug (Pre-IND) meeting is a strategic opportunity that can shape the trajectory of an entire development program....
August 17, 2016
As Phase I clinical trials mark the first time that an investigational new drug is administered to humans, these studies are subject to appropriate current Good Manufacturing Practices (cGMP) in...