
Getting your health technology product to the market is complex and usually consists of several steps that require strategic planning, evidence generation, and stakeholder engagement. Health technology developers (HTDs) that have clear goals set from the beginning, have conducted market research, and have a clear organizational and financial plan, are usually the winning party at market entry and product launch.
Therefore, HTDs that are planning to submit a centralized Market Authorization Application (MAA) to the European Medicines Agency (EMA) should also consider the procedures associated with the recently implemented European Health Technology Assessment Regulation (HTAR). This Regulation will eventually be applicable to all HTDs who plan to prepare a centralized MAA submission to EMA.
The HTAR took effect on January 12th, 2025 for oncology and/or advanced therapy medicinal product (ATMP) therapies. Other health technologies will follow a staggered implementation timeline, with high-risk medical devices and in vitro diagnostics in 2026, orphan drugs in 2028 and all drugs in 2030.
The HTAR covers Joint Clinical Assessments (JCA), Joint Scientific Consultations (JSC), horizon scanning activities, and voluntary cooperation in areas outside the scope of mandatory cooperation, which runs in parallel to the efforts needed to obtain a marketing authorization. HTAR thus requires further organizational efforts from the HTDs, with the internal resources, their skills and roles, being well defined and aligned with the procedures as part of this new Regulation.
.png?width=510&height=647&name=HTAR%20WP_Promo%20Image%20(Blog%20CTA%20Module).png)
Mastering the New European Health Technology Assessment Regulation (HTAR)
Your Complete Guide to Successful Market Access in the EU
Joint Clinical Assessments (JCA) in Europe
A JCA consists of clinical, safety and epidemiological sections that will be assessed jointly by the Member States (MSs) and the European Commission (EC). The HTDs will need to submit a JCA to the EC shortly after their MAA filing to EMA. Subsequently the JCA will be part of HTA decision-making at the national level.
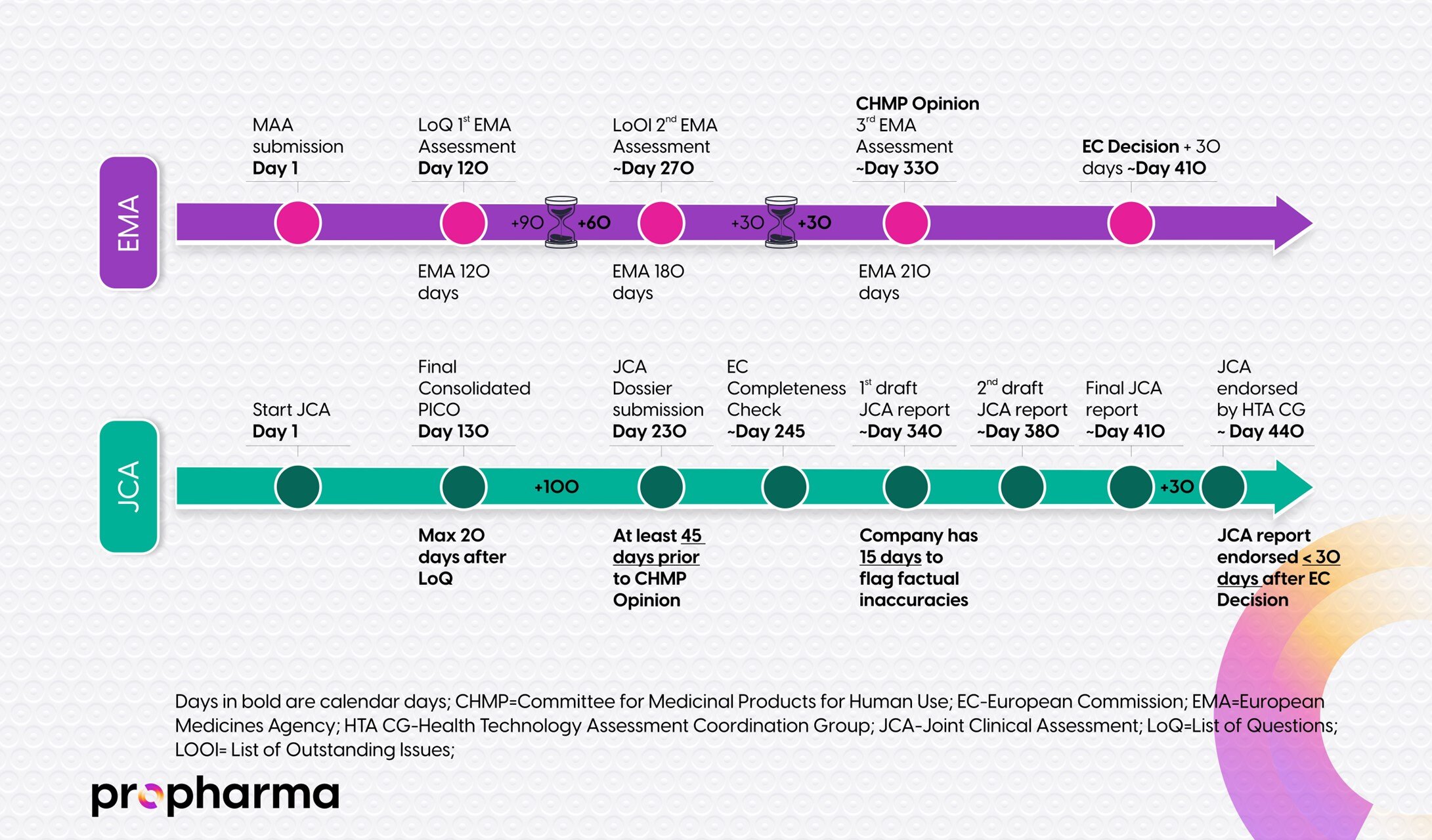
JCA process starts when the MAA dossier is submitted to EMA and the Secretariat of the EU HTA Coordination Group. The EU HTA Coordination Group will assign the JCA Subgroup to review your health technology dossier. Other Subgroups include: the JSC Subgroup, the Methodology Subgroup, and the Identification of emerging health technologies Subgroup. The JCA Subgroup consists of representatives from the member states and is directly responsible for the delivery of the JCA reports. The JCA Subgroup assigns an assessor and a co-assessor of the JCA Subgroup that will compile all the different PICO (Population, Intervention, Comparator(s), Outcome(s)) frameworks from the 27 EU Member States (MS) and present it in a consolidated scoping study that will be shared with the rest of the members of the JCA Subgroup. The consolidated scoping study with the different PICO frameworks deemed relevant to your health technology are then presented to the HTDs. The HTDs will then have 100 days to develop their JCA dossier.
In summary, the JCA steps from the Implementing Act include:
- Start: JCA process starts when the MAA dossier is submitted to EMA and the secretariat of the EU HTA Coordination Group
- The EU HTA Coordination Group appoints the JCA Subgroup
- the JCA Subgroup appoints an assessor and co-assessor from 2 different member states
- PICO frameworks are outlined with involvement of 27 member states
- A consolidated scoping study with the final PICO frameworks are presented to HTD
- JCA dossier preparation by the HTD (100 days)
- JCA dossier submitted by the HTD 45 days before CHMP opinion on the MAA
- JCA dossier evaluated by the JCA Subgroup assessors
- Input stakeholder network (optional and based on availability)
- The EU HTA Coordination Group approves the JCA dossier
- End: Publication by the European Commission < 30 days following the date the European Product Assessment Report (EPAR) has been published by EMA
More details on the timelines for this process are available in Figure 1, below.
How to Prepare for Joint Clinical Assessments (JCA) in Europe
Preparation for the upcoming JCA will help you to create a successful market access plan in Europe, but how can you ensure that your organization is ready to meet all of the requirements?
To this end, ProPharma has developed a four-step process based on what HTAR requires from the HTDs:
Step 1: Introductory Training on HTAR Rules and Implications
To begin the process, introductory training on the HTAR process and its implications is strongly recommended. The trainer will be fully versed and have a complete understanding of the entire process as well as the consequences of noncompliance.
Step 2: Conducting a Technology-Specific Scoping Study
As part of the HTAR process, a scoping study will be conducted by the JCA Subgroup with input from all the EU Member States. As timelines will be short, it is important to be well prepared for the step following receipt of the scoping study by well ahead conducting of a technology-specific scoping study for your product yourself (in collaboration with us) and, based on that, outline a plan for the JCA, including defining the PICOs, before the clock starts.
Step 3: Workshop Preparation to Define Optimal PICO(s)
The finalization of the technology-specific scoping study with PICOs and plan for the JCA dossier development will be arranged in more detail as part of our workshop.
Step 4. Implementation of the Plan
For the final step in the JCA development, the PICO frameworks, as defined by the technology-specific scoping study and the workshop, will be applied, resources added as needed and the timelines as agreed will be adhered to leading to a successful start of the pricing and reimbursement discussions at the national level.
The Value of Joint Scientific Consultations
Prior to MAA and JCA, obtaining early and coordinated advice can improve the chances for favorable pricing and reimbursement outcomes, and thereby facilitating quicker market access. Joint scientific consultations are an opportunity to engage with HTA bodies (HTAb) representatives either alone or jointly with EMA representatives and this process has been implemented alongside the JCA in January 2025. Integrated feedback from both EMA and HTAb will help HTDs to align their development plans with the requirements for marketing authorization and reimbursement.
To qualify for a JSC, the medical product must be likely to undergo a JCA and the request for JSCs must be submitted during designated periods specified in the Annual Work Program of the HTA Coordination Group. Moreover, the request must include all necessary information and evidence to support the consultation, ensuring it aligns with the requirements for a subsequent JCA. Eligibility criteria and selection criteria are at this point in time determined by the number of slots and resources. The JSC Subgroup decides which JSC that are selected.
Joint scientific consultations are strongly recommended to discuss any issues of concern from EMA and HTAb to ensure that HTD’s PICO definitions and trial designs are accurate and can facilitate the development plans.
Expert European Regulatory Affairs and Market Access Consultants with Extensive HTAR Knowledge
ProPharma is uniquely positioned to assist with anything from early regulatory and HTA interactions, to full MAA support with EMA, JCA preparations to the EC, and national HTA preparations and submissions. Our team combines in-house specialists in both regulatory affairs and HTA, enabling us to offer clients unrivalled joint support throughout the entire process. Furthermore, our consultants remain at the forefront of the field by closely monitoring the latest EU guidance and actively engaging with national stakeholders, regulatory bodies, and HTA organizations. Our unique ability to support your team throughout the full product life cycle enables us to help you achieve a successful product launch across the EU and beyond.
Ensure Your Team is Prepared for the Mandatory HTAR
From HTAR preparation to full MAA support and anything and everything in between, ProPharma's team of expert regulatory and HTA consultants is uniquely qualified to help your team achieve successful regulatory and market access interactions.
TAGS: European Medicines Agency (EMA) European Union (EU) Europe General Regulatory Health Technology Assessment (HTA) Regulatory Sciences Market Access

