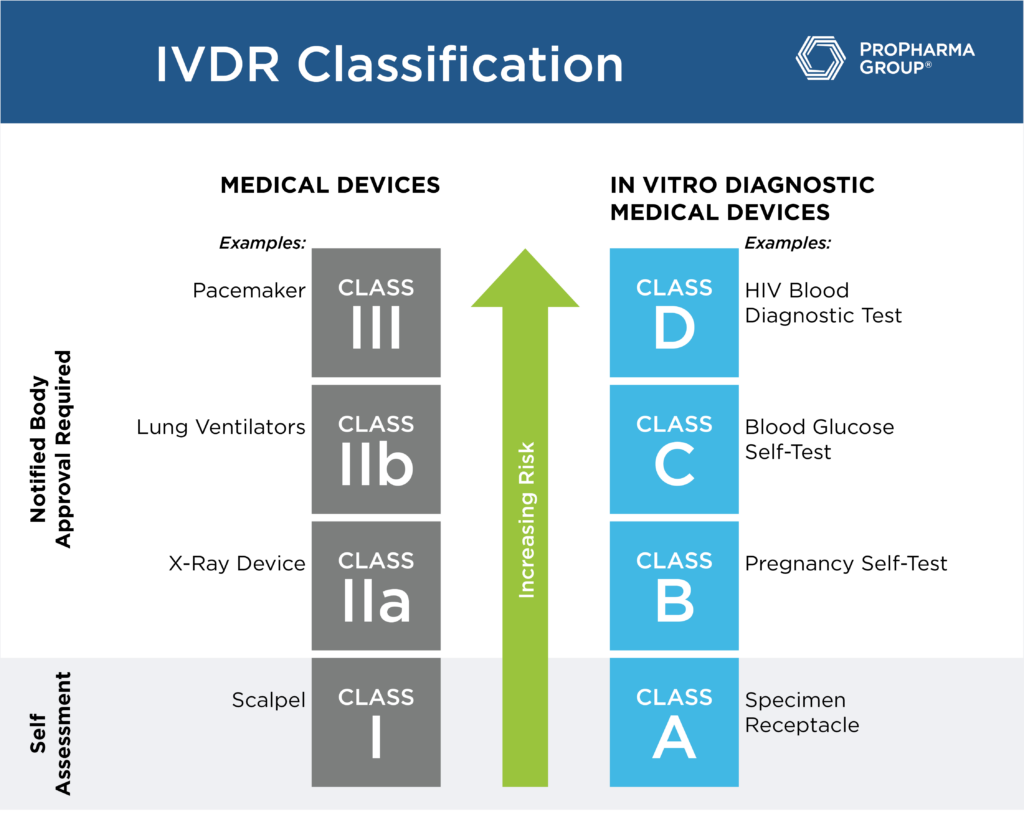
The Medical Devices industry breathed a sigh of relief for the new European Medical Device Regulation (EU MDR), but the May 2021 deadline is now right around the corner. The transition from the current MDD to MDR is not easy, but it will ensure greater transparency so that healthcare practitioners and patients alike can understand and appreciate the safety and efficacy of medical devices.
Many medical devices manufacturers have already put in the hard work and are now on the path toward compliance with the new regulatory framework. Now it’s time to consider both preparing for inspection and, crucially, how to then sustain compliance months after the big change. Here are our tips for managing the next phase toward sustaining MDR compliance.
MDR Certification Preparation Checklist
Internally (outside of your audit group), you should have your own MDC Certification prep checklist to make sure everything is completed. At ProPharma Group, we have a 100-question checklist that we are reviewing with our clients right now to ensure they are fully prepared for inspection. For example, three essential questions are:
1. Has the role of the PRRC (person responsible for regulatory compliance) been clarified?
2. Have the procedures for vigilance and post-market surveillance been clarified?
3. Have the impacted stakeholders all been trained on the MDR and is there awareness across the organization of the MDR and its impacts to medical devices?
Documentation
If you follow good documentation practices then you are well on your way to compliance—but everything needs to be documented from the beginning of the journey when you were doing your gap analysis, to the very end of the journey where you have all your procedures, supporting guidelines templates, roles identified and defined, etc.
Be sure to include decision documents you have developed that capture why you made certain choices. Think about all the key steps that need to be documented. For example, your training records should list all the individuals that were trained, what they were trained on, and when the training occurred. This way, all of your documentation will be together and readily available for when you need it. The goal here is to make life as easy as possible for the Notified Body that will be reviewing your materials.
Economic Operators
Make sure that your Economic Operators, such as distributors, manufacturers, and suppliers, are all planning to move to MDR and that they are in the process of or have completed their remediation work. Be sure you understand their classification of the devices (in case of device classification changes) and review and understand how and when they plan to complete the MDR certification process. You may need to align timing or, at least, speak to it with your Notified Body. Not every partner will decide to go through the effort to become MDR certified. If that happens, a critical and important goal is quickly determining and implementing a mitigation strategy.
Sustaining Long-Term Compliance
Although it is tempting to focus solely on upcoming inspections and the May deadline, it is crucial to also start preparations that will help sustain compliance months after certification. Look ahead to a year from now and think about what’s needed to remain compliant after the big deadline. We suggest focusing on four main areas:
EUDAMED: Health care practitioners and patients will be able to use EUDAMED to access information about the device, safety, and clinical performance data. Though implementation begins in December 2021, Manufacturers will need to work on this past May 2021, making it a continued area of focus. Make sure that you have a team actively working on EUDAMED to systematically implement all of the different requirements as the modules continue to roll-out.
Oversight for new processes: After you have updated your processes for MDR, make sure that you have people in place to drive all these processes. You will need to have proper oversight to make these new processes run effectively. Set up proper sequencing of the process outputs as some are dependent on others. Establish a governance group that will discuss any issues or risks that result due to trying to execute or implement all these new or updated requirements.
Communicate new guidance: There will certainly be additional guidance coming from the MDCG (Medical Device Coordination Group) or other related groups in the future, so be sure there is a dedicated Regulatory Intelligence team in place. Create a plan so that guidance will cascade to the impacted stakeholders and they will review the new guidance to figure out how to best address it, if applicable.
Manage compliance: Conduct internal audits to make sure that your team is still maintaining compliance to MDR long after the rush toward the deadline. The same holds true for your Economic Operators. Having an internal regulatory memory will help keep the pressure consistent, both internally and with your partners.
The Patients are the End Goal
The most important reason to put effort into maintaining compliance is the patients. All of the hard work put into a remediation of this scale will help ensure that patients get the medical treatment they need and the transparency to make informed decisions and understand medical devices.
There will be a lot of interest and excitement around being able to actively pull information about devices and understand their efficacy and any potential risks. If any problems arise with a specific lot, there will be better traceability so that any issues can be addressed much faster and more effectively in the future.
Any transition to a new regulatory framework is a large undertaking, but you don’t have to do it alone. Our experts can help guide you in the right direction either directly for project support and/or with our webinar series that covers a range of topics focused on helping manufacturers efficiently transition from the current MDD/IVDD regulatory framework in Europe to MDR/IVDR.