
Implementing the eSTAR Format
The eSTAR template is a positive step for both CDRH and medical device Sponsors; but, as with any new tool, there are challenges to utilizing the template. Sponsors need to adapt the way they create and compose 510(k)s because the eSTAR software presents different challenges compared to FDA's old eCopy format.
An important adjustment will be for Sponsors to develop a collaboration plan as the eSTAR template is not set up to handle multiple users, editing within the template, or tracked changes, and becomes cumbersome once attachments are added. Keeping track of the multitude of attachments and references to specific information they contain is also a new challenge when using the template.
ProPharma can ensure your team has a smooth transition to filing 510(k) submissions in FDA's eSTAR format so your regulatory team doesn’t have the added stress. In addition to technical expertise, we offer project management capabilities that simplify authoring, reviewing, and finalizing FDA submission content.
Continual Improvements to eSTAR
CDRH has released updates to eSTAR and is currently using Version 5.2 of the 510(k) eSTAR template, which was released on March 29, 2024. CDRH releases a major revision when there are policy or regulatory changes.
For example, a major revision from 3.0 to 4.0:
4.0 (2023-06-12): Software section updated according to FDA Software Guidance document
And a minor revision from 4.1 to 4.2:
4.2 (2023-09-11): Resolved bug preventing Additional Information responses from being deleted
As Sponsors assemble their submissions, it is important to stay up to date on which major version CDRH is using and what expectations have changed from the last version. Submitting a 510(k) Premarket Notification to FDA using an outdated version may result in a submission with incomplete information and requests from the CDRH review team for additional information, which may delay the regulatory review.
ProPharma team of expert regulatory consultants tracks all revisions, both major and minor, to ensure there are no technical issues with the eSTAR template and that no critical information is omitted. This allows your regulatory team to focus on content of the submission and be assured that technical aspects are not being overlooked.
Product design, testing, and manufacturing are all complete – you’re ready to send your data package to FDA for clearance so you can begin marketing your medical device. The last hurdle is compiling a complete and technically correct 510(k) submission so that the regulatory review is not delayed by an Additional Information Request (AI Request) or Refuse to Accept (RTA) letter. When it comes to 510(k) submissions, eCopy is out and eSTAR is in, along with a new set of rules to follow. Is your team up to date on how to submit a 510(k) to FDA?
What Are CDRH’s eSTAR Templates?
To streamline the medical device review process, FDA’s Center for Devices and Radiological Health (CDRH) has invested in developing interactive templates for Sponsors to use for submission of their 510(k) Premarket Notifications, Premarket Approval (PMA) submissions, De Novo Classification Requests, and Pre-submission Meeting Requests. While use of the templates remains optional for PMAs, De Novo, and Pre-submissions, as of October 1, 2023, use of FDA’s eSTAR template is now required for 510(k) Premarket Notifications.
How does the FDA eSTAR Template Help Sponsors?
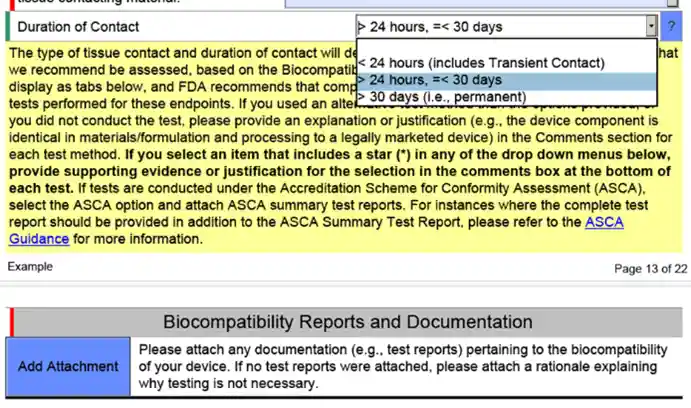
The eSTAR templates have built in prompts that are intended to assist the Sponsor with providing all of the information FDA’s team will need to review the product. The idea is to eliminate Refuse to Accept (RTA) responses because the Sponsor will have been instructed to attach or include pertinent information with their original submission. The image below shows an FDA prompt for additional information related to biocompatibility testing in the 510(k) eSTAR template.
How does the eSTAR Template Help FDA?
With 19,100 submissions received in 2023 (CDRH 2023 Annual Report), it makes sense that CDRH designed templates to help increase the efficiency of its review process. The eSTAR templates follow FDA’s internal review checklists, standardizing Sponsor applications and eliminating the need to search for information within a submission package.
ProPharma: Expert 510(k) Submission Consultants
ProPharma helps clients turn creative ideas into marketed medical devices while ensuring compliance with the complex array of laws and regulations in the US and internationally. We provide a wide range of services to assist in the development of novel medical devices, including medical equipment, implants, in vitro diagnostics, imaging, companion diagnostics, mobile and analytical software devices, combination products, laboratory-developed tests, and drug delivery devices. In addition to technical eSTAR expertise, we help Sponsors formulate effective development strategies, determine the optimal regulatory pathway for their product, plan and manage key projects, prepare and submit US and European regulatory filings, and manage regulatory affairs in the US and internationally. We can help design and implement quality management and risk management systems, provide guidance on CLIA regulations, conduct GMP and GCP audits to ensure regulatory compliance, and address critical issues that arise during development programs.
We have unsurpassed knowledge of the product development and regulatory environment and are thoroughly familiar with the review expectations of FDA’s CDRH. By utilizing the best talent available anywhere in the world, ProPharma delivers extraordinary value to its clients.
TAGS: Food & Drug Administration (FDA) Medical Devices General Regulatory FDA 510(k) FDA Submission Regulatory Sciences

