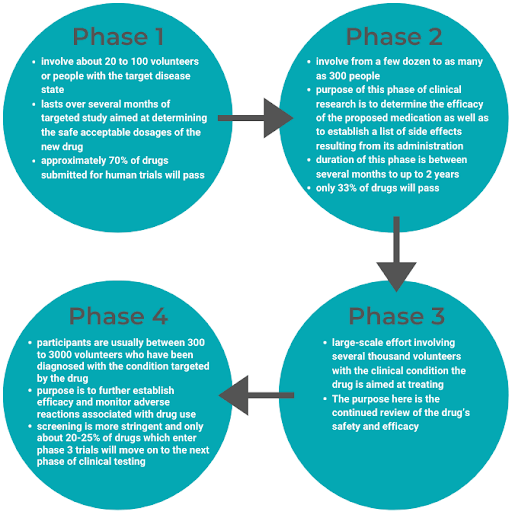
Congratulations, FDA has approved your PMA! Now what?
Securing a Premarket Approval (PMA) from the FDA is a significant achievement for any medical device manufacturer—a validation of your product's safety and effectiveness. But the journey doesn't end there. The real challenge begins post-approval, where careful management of your PMA is crucial to ensure ongoing compliance, maintain market access, and safeguard your product's reputation. In this blog, we'll discuss the proper "care and feeding" of your PMA, exploring best practices for managing FDA expectations, controlling processes, and navigating the complexities of changes and updates. Through real-world examples, we’ll illustrate how to stay ahead of the curve and keep your PMA in good standing.
PMA Conditions of Approval
When your medical device gets approved by the FDA, it includes “Conditions of Approval”, which are specific requirements from the FDA that the Sponsor must adhere to. Common examples of PMA Conditions of Approval can include post-approval studies, annual reporting, labeling requirements, device tracking and traceability, and more. These conditions of approval are designed to ensure that the device continues to meet the FDA's stringent safety and effectiveness standards even after it has been approved and is in use in the market.
It is important to remember that the PMA Conditions of Approval as a written contract with the FDA; should this contract fail to be met, the PMA can be revoked. Where the PMA Sponsor is unable to meet the requirements, additional communication with FDA to get agreement on changed conditions of approval should be completed as soon as is discovered.
Post-Approval Studies as part of PMA Conditions of Approval
Most PMA Conditions of Approval include requirements for Post-Approval Studies.
PMA Annual Reporting Requirements
Separate from the Annual Post Approval Study reports, the PMA Annual report is submitted at intervals of 1-year from the date of the original PMA approval order (unless otherwise agreed to by FDA). The report provides the FDA with updates on the device’s performance, any changes made, and other relevant information over the past year.
By reviewing these reports, the FDA can assess whether the device continues to meet safety and effectiveness standards, identify any potential issues early, and take appropriate action if necessary. Failure to submit a PMA Annual Report or to include all required information can result in regulatory actions, including the revocation of the PMA, which would prevent the device from being marketed or distributed.
Key Components of a PMA Annual Report
Cover Letter
Every annual report will have a Cover Letter that includes:
- PMA Number # – a unique identifier assigned to the device’s PMA
- Device Name – the commercial name of the device
- Company Name – the name of the manufacturer or sponsor
- Date of Report – the date the report is submitted
- Reporting period – the time frame covered by the report, typically one year from the data of the original PMA approval or the anniversary of the previous annual report
Changes made during the Reporting Period
During the year, a chronological list of changes to the product design, manufacturing, supplier changes, labeling updates and other changes is maintained by the PMA Sponsor. Maintaining a file structure for each individual change is recommended for ease of compiling the PMA Annual Report. Some changes related to clarification, correction of typos and translation of requirements from a drawing to a specification may not require notification in the PMA Annual Report. The PMA Sponsor’s QMS should have a clear decision tree for determining what changes should be included in the PMA Annual Report.
Include any supplements or 30-day Notices and document the document number of the submission and the status of the submission (approved/date or pending).
For changes that did not rise to the level of a PMA Supplement or a 30-Day Notice, FDA recommends presenting these changes in the PMA Annual Report in separate tables for manufacturing changes, design changes and labeling changes. It is appropriate to document linkages between each of the tables for changes that are associated with each other.
Recommended table format (example)
Suppliers involved with the manufacturing of PMA product
It is the responsibility of the PMA Sponsor to ensure that the PMA Section 4 Design and Manufacturing of the original PMA submission continues to be valid. In practice, this includes Supplier Quality Agreements that require notification of planned changes to the PMA Sponsor for review for Regulatory requirement for submission of supplements or 30-Day Notices.
The Supplier Quality Agreement includes the requirement for maintaining the annual FDA Establishment Registration for the Supplier facility and the agreement to allow FDA to inspect the supplier’s facilities.
We recommend that Supplier Quality Agreements include the specific list of changes that require notification, such as use of new equipment, additional processing steps, new chemicals for cleaning, move of equipment, move of facility, substitution of materials, etc. for clarity for suppliers. For PMA products, any change is scrutinized for requirement for a PMA supplement or 30-day notice.
Changing suppliers from those that were in the original PMA submission requires a PMA supplement and potentially an FDA inspection of the new supplier. Changes of suppliers should be carefully planned as FDA review of the supplement and the FDA inspection could take up to 12 months.
Design Changes
For design changes, FDA recommends that all relevant reasons for the change be documented in the PMA Annual Report, such as:
- Device improvement or enhancement
- As a result of an adverse event, device defect or failure reported to the PMA Sponsor, or as a result of literature review. If there were any reportable events to FDA, a mention of the eMDR# and UDI
- Changes made in response to a complaint, request or suggestion.
- Changes associated with recall or corrective action, a mention of the recall # or if a Class III recall, identify internal #.
- Changes related to an FDA Safety alert, Warning Letter, or Public Health Notification
- Changes related to Advisory Notices being sent to practitioners, patients, or distributors.
All design changes require a robust justification for why the change does not adversely impact the safety and performance profile of the original PMA submission device. Adding a size outside of the approved device range requires pre-clinical testing to be submitted as part of the PMA Supplement to document the safety and performance profile is not changed as compared to the original PMA submission device.
Labeling changes
The best way to think about labeling is "FDA owns the labeling for PMA products". The PMA Sponsor must evaluate labeling changes with the PMA in mind. Changes in labeling to add warnings or contraindications may require FDA review and approval.
Summary and Bibliography of report of Scientific Investigations and Literature
Summary and Bibliography of report of Scientific Investigations and Literature is compiled by the PMA Sponsor for articles/reports not submitted in the original PMA submission. The summary includes all unpublished reports of data from other clinical investigations involving the PMA device or related devices. As part of the summary, a discussion of how conclusions from the reports/literature impact the safety and performance profile of the PMA product.
Complaint listing for the period of the PMA Annual Report
A full listing of complaints related to the PMA Submission is included along with any conclusions and any eMDRs as referenced by the MDR# and UDI. A summary statement describing the complaints and resolutions is recommended. If there were any recalls or corrective actions, a table with recall dates and status is recommended.
Information on Devices Shipped or Sold during the Reporting Period
A complete listing of all products with part numbers, lot numbers, descriptions, quantities, locations used, date used, and full string Unique Identifier is documented in a table for the reporting period.
FDA Review of PMA Annual Reports
FDA completes the review of the PMA Annual Report and documents the following:
- An acknowledgement of the Annual Report – FDA will provide an email or letter documenting that no additional information is needed.
- Request for Additional information – The FDA does not find that the information provided is sufficient and requests additional documentation via letter or email.
- Changes not appropriate for an Annual Report – FDA determines that changes require a supplement or a 30-Day Notice.
Key Takeaways for Effective PMA Management
Navigating the post-approval landscape of a PMA is no small feat. It requires diligent oversight, meticulous record-keeping, and a proactive approach to managing changes and communicating with the FDA. By adhering to the guidelines and recommendations discussed in this blog, you can ensure your PMA remains compliant and your device continues to meet the highest standards of safety and efficacy. Remember, the key to successful PMA management lies in treating it as an ongoing partnership with the FDA, where transparency and adherence to agreed-upon conditions are paramount.
Work with ProPharma
Need expert guidance on managing your FDA PMA? ProPharma is here to help. Our team of medical device regulatory experts and industry veterans can assist you with everything from initial submissions to post-approval management, ensuring your device remains compliant and market ready. Contact us today to learn how we can support your PMA and help you navigate the complexities of FDA regulations.