June 28, 2021
June 28, 2021
In late 2018, FDA announced changes to modernize and increase the efficiency of its 510(k) clearance pathway, which is the most common medical device submission and allows for the allows for the "clearance of low- to moderate-risk devices that are substantially equivalent to a device already on the market – otherwise known as a predicate device." Since this announcement was made, FDA has taken a number of actions to bring efficiency to the process. These changes have also helped the Agency keep pace with evolving technology and reflect the advances in the safety and capabilities of a new generation of medical devices. The FDA believes that "new medical devices that come to market under the 510(k) pathway should either account for advances in technology or demonstrate that they meet more modern safety and performance criteria".
FDA still firmly believes in the merits of the 510(k) process, but explicitly stated that innovative advances in technology have forced the Agency to evolve and to anticipate upcoming technological changes.

The Medical Device Amendments of 1976 established three categories of medical devices: Class I, II, III. Each class is based upon the level of risk the device poses to the user.
Included in the amendment was the provision for the 510(k), which specified that if a new device was substantially equivalent (SE) to a device legally marketed in the US before May 28, 1976, it "could proceed to market with the same classification and controls".
To obtain substantial equivalence, the devices in question are not required to be identical. SE is established with respect to; "intended use, design, energy used or delivered, materials, performance, safety, effectiveness, labeling, biocompatibility, standards, and other applicable characteristics".
In 2020, FDA’s Center for Devices and Radiological Health (CDRH), cleared 2,901 devices through the 510(k) pathway. In the recent past, CDRH has made efforts to improve the performance, predictability, efficiency, and safety of the 510(k) program. FDA believes that its recent improvements will continue to assist its efforts to encourage innovation, promote modern patient care, and match its evolving understanding of benefits and risks. The Agency states that the most impactful way to achieve its goal is to encourage innovators to rely on more modern predicate devices or objective performance criteria when seeking to bring new devices to patients.
The devices reviewed through the 510(k) program are growing increasingly complex. They are smaller, more portable, and are more interconnected. Technology has changed the way healthcare providers and patients interact with devices, and all of this has prompted the need for guidance.
In 2019, the Agency issued final guidance establishing an alternative pathway which expanded the concept of FDA’s Abbreviated 510(k) pathway. This guidance, entitled "Safety and Performance Based Pathway," allows manufacturers of certain, well understood types of medical devices "to use FDA-identified performance criteria to demonstrate that a device is as safe and effective as a predicate device." The goal is to finalize the pathway to "expand its use broadly across the 510(k) program and make it the primary pathway for devices eligible for 510(k) review."
On January 6, 2021, FDA finalized its 2019 draft guidance entitled "Safer Technologies Program for Medical Devices." "The Safer Technologies Program (STeP) is a voluntary program intended to decrease the time to market for medical devices or device-led combination products that are "expected to significantly improve the safety of currently available treatments or diagnostics that target an underlying disease or condition associated with morbidities and mortalities, less serious than those eligible for the Breakthrough Devices Program." The goal for the program is to expedite the development, assessment, and review of these products, without compromising the FDA’s statutory standards.
These changes to the 510(k) pathway are just two of the ways FDA is working to bring life-saving devices to market as quickly as possible while also keeping up with modern technology. With these new innovations, the Agency’s top priority remains to assure the safety and effectiveness of devices for patient safety.
Are you in the process of developing a medical device? We can help ensure that your 510(k) is compliant with FDA’s changing rules and regulations. To learn more about our services and how we can help with your medical device submission, contact us today.
TAGS: Regulatory Sciences

August 15, 2022
Commercializing your medical device in the US market often requires submitting a marketing application to the FDA to become an FDA Approved or Cleared Medical Device. The content of your FDA...
April 3, 2018
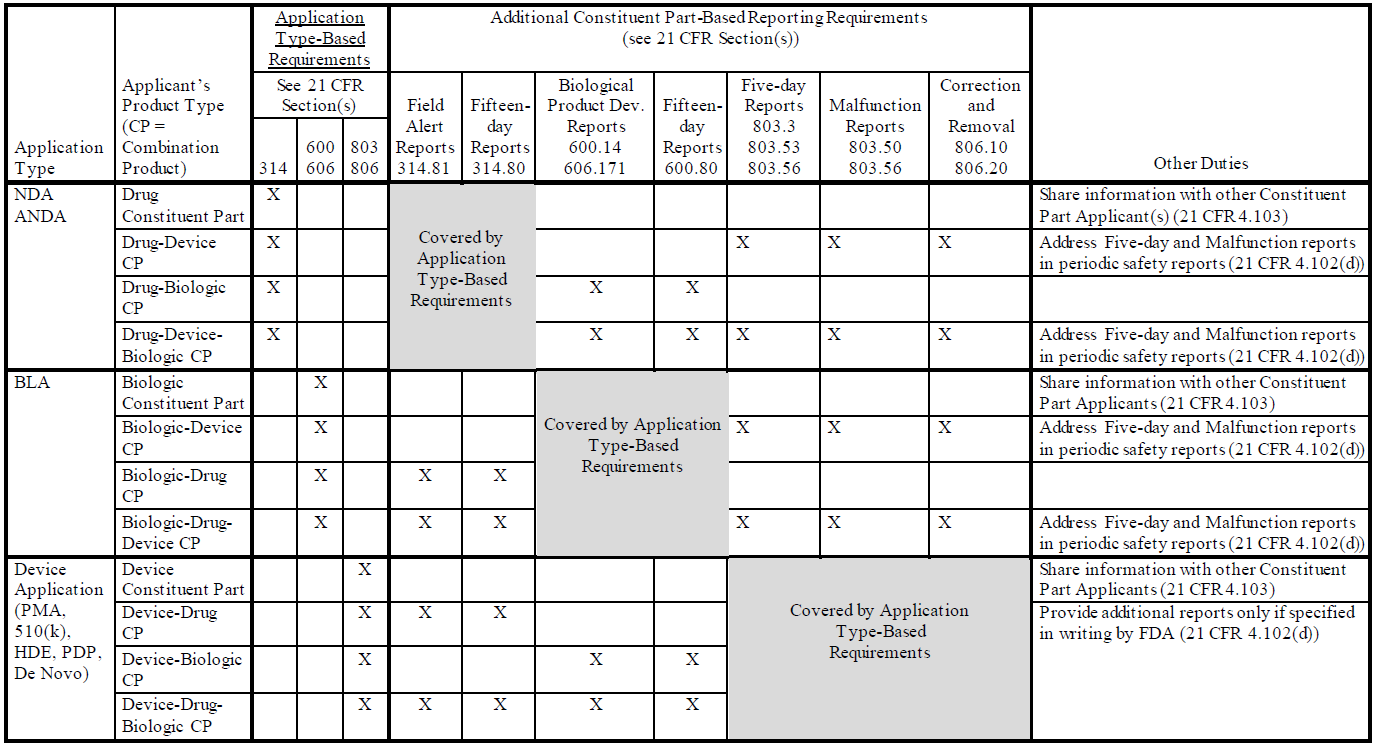
On March 20, 2018, the US Food and Drug Administration (FDA) released two new guidance documents to help companies comply with the December 20, 2016 final rule establishing postmarketing safety...

January 9, 2019
From FDA's approval of the first cannabis-based product in the U.S., to the classification of two Apple Watch apps and changes in the Agency's submission requirements related to Sponsor meetings,...